ISO 14971 for medical device risk management was approved in December 2019. Although no significant changes on the risk management process was defined, a substantial re-organization of the standard was performed.
Click here for a Risk Management Procedure aligned with the new ISO 14971:2019.
The main changes on the version 2019 of ISO 14971 includes new terms and a more detailed requirements on post-market risk management
One big substantial change is related to the annexes of the standard. This last revision contains only three annexes, whereas the remaining ones have been moved to ISO 24971, which has been published in 2020; we have been discussing in this post about ISO 24971. Specifically, the annexes that remained within ISO 14971 are:
- rationale for requirements,
- risk management process,
- and fundamental risk concepts.
It is expected that ISO / TR 24971 will become essential for risk management for medical devices and it will contain all the annexes which are not currently present on ISO 14971.
One of the key activities related to risk management is the risk analysis. There are different methodologies to perform a risk analysis, one of them is the so-called FMEA – Failure Mode Effect Analysis.
General Overview of the ISO 14971:2019
Particular attention was paid on the newly updated ISO 14971:2019 on the benefit-risk analysis of medical devices, so to align the standard with EU MDR (2017/745) and IVDR (2017/746). The new ISO 14971 now requires to perform an assessment of overall residual risk and to determine the criteria for risk acceptability. The methodology to assess the acceptability of the overall residual risk can be different from the acceptability criteria of individual risks.
New terms and definition was also added in the new standard, including benefit, state of the art and reasonably foreseeable misuse.
Important updates was given on cybersecurity side, reinforcing the importance to evaluate the security-related risks that come from connected devices. This attention to cybersecurity is aligned with FDA and other regulatory agency behavior, that in last years have increased the focus on medical device cybersecurity.
Overview of the Risk Management Process
The overall risk management process can be described by the scheme below:
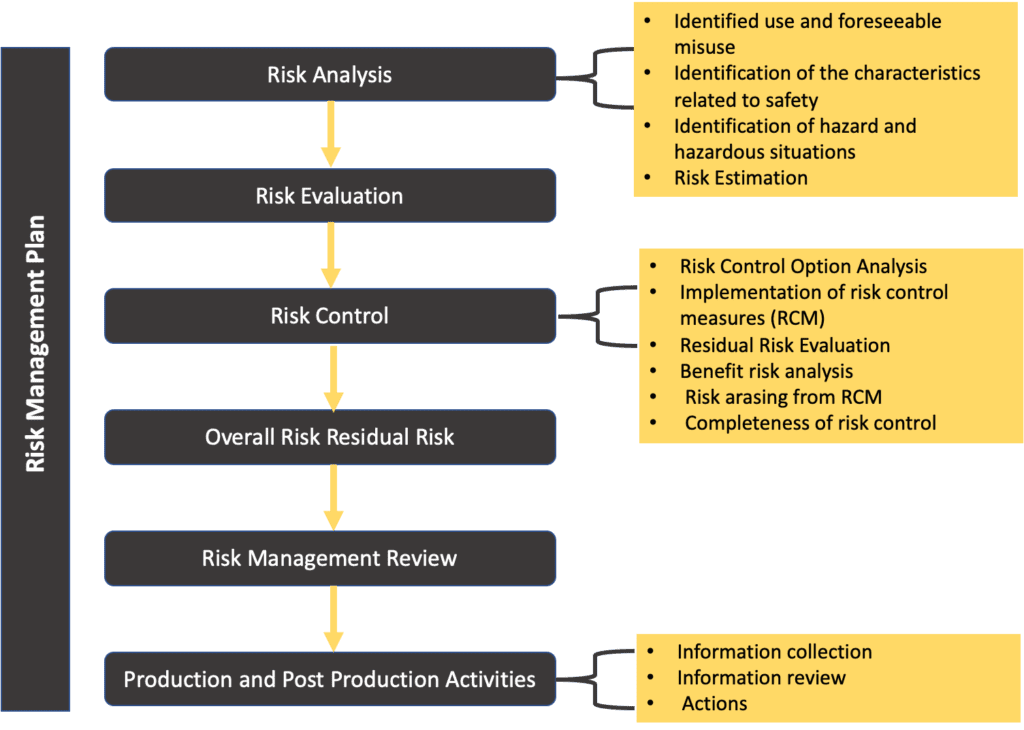
Basically, the risk management process has the goals of
- identification of hazards and hazardous situation
- estimation and evaluation of the risks
- Risk control
- monitoring and effectiveness of the risk control measures
Risk Management Plan
The risk management plan is one of the most important document of the risk management process. Here below, we summarise within the following table the main contents of the risk management plan:
| Content of the Risk Management Plan |
|---|
| Scope of Risk Management Activities |
| Responsibilities and Authorities |
| Requirements for review of Risk Management Activities |
| Criteria for Risk Acceptability |
| Method for evaluation of the residual risk |
| Methods for verification of risk control measures |
| Post-production risk management activities |
Risk Control according to ISO 14971
Risk Control Measures
We are going to talk about only few specific steps of the risk management process. One of them is the Risk Control part of the process.
Risk control options are of fundamental importance in order to reduce the risks. It is essential that risk control measures are going to be implemented following a specific priority order:
- inherently safe design and manufacturing of the device
- protective measures in the medical devices itself or in the manufacturing process
- information for safety and / or training;
When risk reduction through implementation of risk control measures are not feasible, a benefit risk analysis shall be performed and the residual risk shall be evaluated and discussed.
Verification of the risk control measures
All the risk control measures which are identified need to implemented and verified. The type of verification performed depends of course on the nature of the risk control measures; typically it can be done through a specific tests, visual inspection, validation activities, etc. It is possible, of course, to combine verification activities conducted in the framework of the design process with verification of the effectiveness of the specific risk control measure.
Residual Risk Evaluation
After the implementation of the risk control measure, the residual risks shall be evaluated by comparing it with the risk acceptability threshold defined in the risk analysis.
It is important to mention that any type of risks need to be reduced as far as possible, including risks that by nature are relatively low. In any case, after the implementation of risk control measures, it is not possible to have any unacceptable risks defined in the risk analysis. If, during lifetime of a device, an unacceptable risk came up, actions on the field (recall, safety notice) shall be implemented to immediately reduce this risk to an acceptable level.
Benefit-Risk Analysis
In case a residual risk is not evaluated as acceptable, a benefit-risk analysis shall be documented to demonstrate that the benefits of the intended use outweight this residual risk.
Risks arising from risk control measures and review of risk control measures
The effect of risk control measures shall be reviewed to evaluate whether new hazards have been introduced and if the risk control measure affects the estimation of the risks for previously identified hazardous situations.
Moreover, the risk control activities shall be reviewed to make sure that these activities are competed and all the risks associated to the identified hazardous situations have been identified.
Conclusions
The most important updates on the newly ISO 14971 is the post-market risk management section. Specifically clause 10 of the standard have been renamed Production and post-production activities and it is now more aligned with Clause 8 of ISO 13485. Clause 10 highlights the necessity of an active process for post-market risk management. It establishes a system to collect production and post-production information and evaluate this information from risk point of view. A very interesting document on postmarked risk management is the one published by AAMI .
In conclusion, the updated ISO 14971 for medical device risk management along with New ISO 20471 on labelling requirements will become important tools for Medtech companies to foster product safety and regulatory compliance.
QualityMedDev Risk Management Documentation
Nobody can deny the importance of risk management in the medical device field. The regulation in the last 10 years shifted completely towards a situation where risk management process is at the core of quality management system and technical documentation for medical devices. In order to support the implementation of an efficient risk management process, QualityMedDev provides different documentation which can definitely help your organization in the implementation, reorganisation or improvement of risk management. In fact, in the QualityMedDev DocShop, the following documentation can be downloaded:
- Risk Management Procedure , to ensure that the risk management process is well defined within your Quality Management System
- Risk Management Plan Template , which can be used as starting point for the practical implementation of the risk management process
- Risk Analysis Template, which provides you a great example of template that can be used for your risk analysis.
Moreover, QualityMedDev has recently published an e-book focused on risk management process for medical device sectors. Check it out here below and fell free to download it !
Subscribe to 4EasyReg Newsletter
4EasyReg is an online platform dedicated to Quality & Regulatory matters within the medical device industry. Have a look to all the services that we provide: we are very transparent in the pricing associated to these consulting services.
Within our WebShop, a wide range of procedures, templates, checklists are available, all of them focused on regulatory topics for medical device compliance to applicable regulations. Within the webshop, a dedicated section related to cybersecurity and compliance to ISO 27001 for medical device organizations is also present.
As one of the leading online platforms in the medical device sector, 4EasyReg offers extensive support for regulatory compliance. Our services cover a wide range of topics, from EU MDR & IVDR to ISO 13485, encompassing risk management, biocompatibility, usability, software verification and validation, and assistance in preparing technical documentation for MDR compliance.
Do not hesitate to subscribe to our Newsletter!
