Introduction
In the last years, the use of Artificial Intelligence and Machine Learning software as Medical Device has increased substantially. On the other hand, the regulators struggled to adapt the Medical Device Regulation to this new type of devices.
Recently, an interesting article has been published on Digital Medicine: The need for a system view to regulate artificial intelligence/ machine learning-based software as medical device where the obstacles in the adoption of the current FDA regulations for AI/ML medical device is well described.
One of the first medical device based on Artificial Intelligence which is able to perform a diagnosis was IDx-DR, an AI diagnostic system that detects signs of diabetic retinopathy in retinal images.
Challenges for Regulatory Compliance
One of the biggest problem from regulatory point of view is the control of the change of the device. A machine learning device should in theory work as a continuous improved systems; basically the most valuable asset of AI/ML is its ability to improve by learning from data by using the so-called adaptive algorithm. On the other hand, FDA and similar regulators require a strict control on the changes of devices (especially if these changes have impact on the performance of the device). Thus, it is possible that modification of AI/ML system will require new FDA approval.
FDA Analysis on the Possible Changes for AI/ML-Based Devices
In this contest, FDA recently published a discussion paper (and request for feedback) related to a proposed regulatory framework for (AI/ML)-Based Software as a Medical Device.
In this discussion paper, the agency envied different types of possible modification for an AI/ML system:
- Performance – clinical and analytical performance;
- Inputs used by the algorithm and their clinical association to the SaMD output; and/or
- Intended use – The intended use of the SaMD, as outlined above and in the IMDRF riskcategorization framework, described through the significance of information provided by the SaMD for the state of the healthcare situation or condition.
To tackle this problem, as first step the agency suggest the implementation of a Total Lifecycle Regulatory Approach which can be summarised in the scheme below:
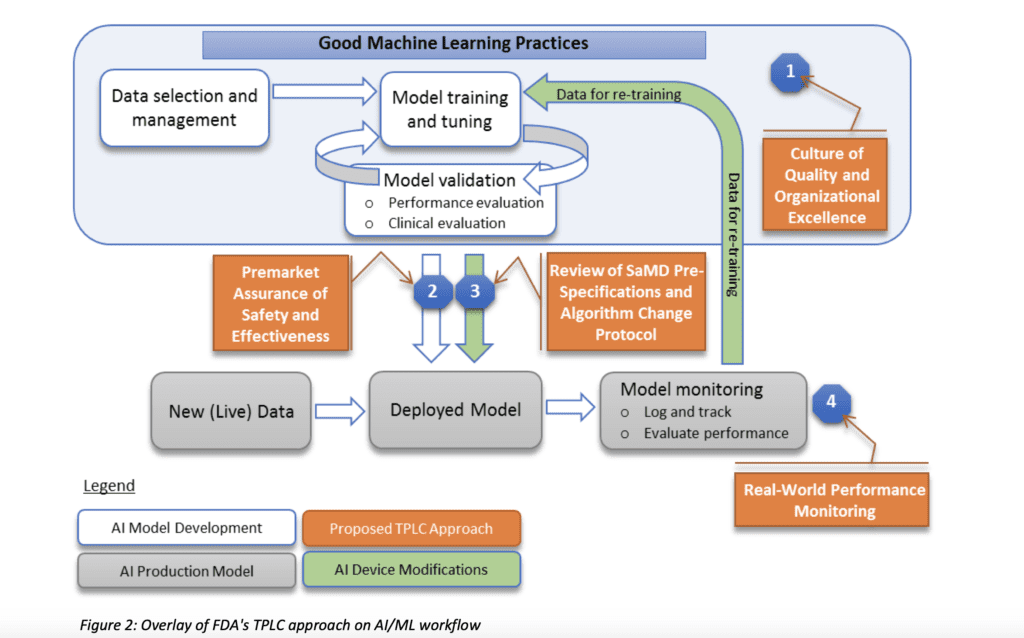
The key concepts in this model are 1) incorporation of risk management that will drive the decision wether a change in the software requires a 510(k) application 2) the surveillance on the performance of the software based on real works data. Other important factors are as well:
a) ensure to have clear expectations on quality systems and good ML practices (GMLP).
b) perform premarket review for those SaMD that require premarket submission to demonstrate reasonable assurance of safety and effectiveness.
Let’s concentrate on the approach suggested by the agency to manage software modification after the market approval. As we know, manufacturers need to evaluate the modifications based on potential risk to patients. The software modifications guidance uses a risk-based approach and expects a manufacturer to perform a risk assessment and evaluate that the risks are reasonably mitigated. The outcome could be either a new 510k application or documentation of the change and analysis from risk point of view.
FDA Proposal on how to handle changes for AI/ML-Based Devices
To understand how the agency is willing to regulate modifications of AI/ML systems, it is needed to introduce the concepts of predetermined change control plan, which is basically constituted by two factors:
SaMD Pre-Specifications (SPS): A SaMD manufacturer’s anticipated modifications to “performance” or “inputs,” or changes related to the “intended use” of AI/ML-based SaMD. The SPS draws a “region of potential changes” around the initial specifications of the original device.
Algorithm Change Protocol (ACP): Specific methods that a manufacturer has in place to achieve and appropriately control the risks of the anticipated types of modifications delineated in the SPS.
The Ai/ML-based software modification will be handled according to the scheme below:
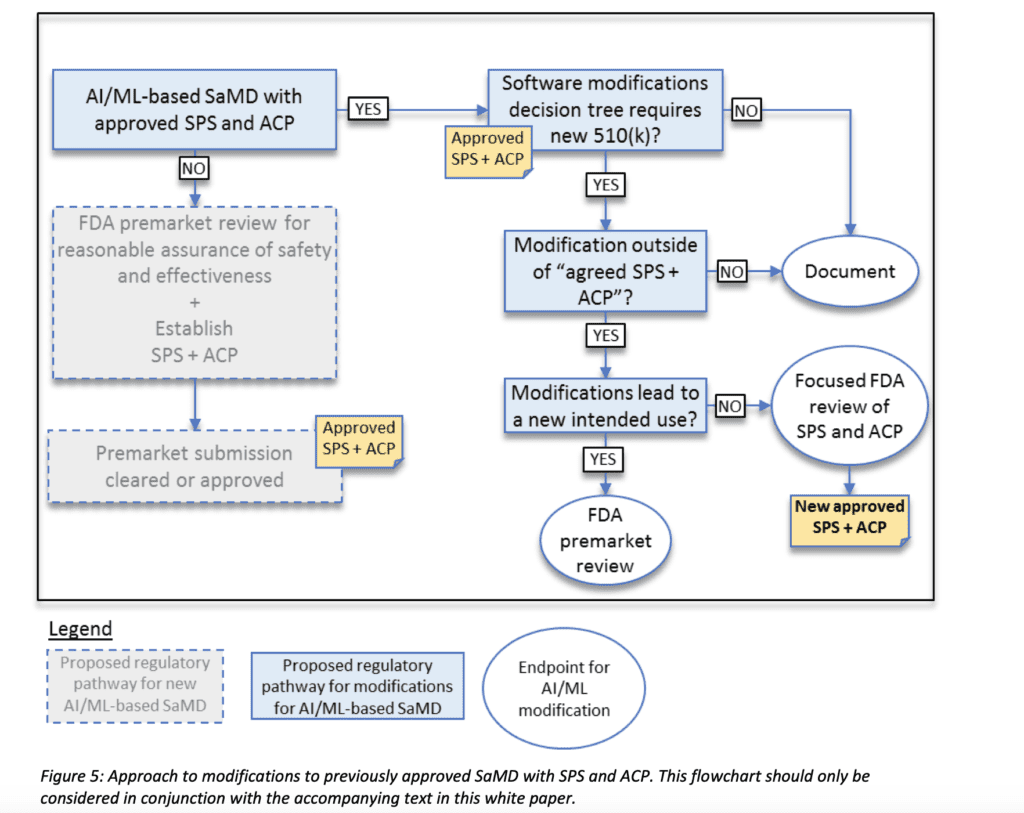
So basically if the proposed change stays within the planned SPS/APC, no new 510(k) application would be required.
This approach would definitely be more flexible and suitable for AI/ML-based system which, by nature, require modifications during their product life-time.
It is important to state that for the moment this is just a proposal of regulation for Ai/ML products. This proposal is open for discussion and feedback has already been published on the web.
Subscribe to 4EasyReg Newsletter
4EasyReg is an online platform dedicated to Quality & Regulatory matters within the medical device industry. Have a look to all the services that we provide: we are very transparent in the pricing associated to these consulting services.
Within our WebShop, a wide range of procedures, templates, checklists are available, all of them focused on regulatory topics for medical device compliance to applicable regulations. Within the webshop, a dedicated section related to cybersecurity and compliance to ISO 27001 for medical device organizations is also present.
As one of the leading online platforms in the medical device sector, 4EasyReg offers extensive support for regulatory compliance. Our services cover a wide range of topics, from EU MDR & IVDR to ISO 13485, encompassing risk management, biocompatibility, usability, software verification and validation, and assistance in preparing technical documentation for MDR compliance.
Do not hesitate to subscribe to our Newsletter!
