The complaint handling process for medical device manufacturers is of fundamental importance for multiple reasons. Surely, compliant process is directly linked with the safety of the devices on the field and, in terms of quality management system, linked with other processes such as risk management, vigilance reporting and other active post-market surveillance activities.
We have been already talking about the vigilance reporting requirements according to the European Medical Device Regulations and according to FDA regulation (21 CFR 803). In this article, we want to give a more high level overview of the whole complaint process, from the reception of a complaint until its closure.
What is a Complaint
FDA provides a precise definition of complaint in the section 21 CFR 803 (b):
Any written, electronic, or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, safety, effectiveness, or performance of a device after it is released for distribution.
As you can see, the key characteristic for a complaint is the presence of an allegation against a specific device. This is very important as it will be useful to determine if a specific communication received from the customer can be considered a complaint or not.
Overview of Complaint Handling Process
The process for complaint handling can be summarised in the scheme below.
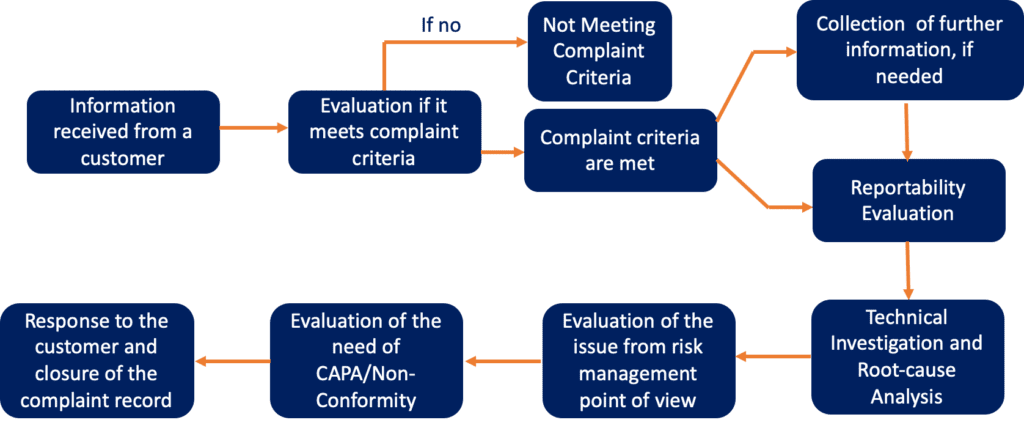
What type of information need be mentioned in a complaint received from a customer?
When a complaint is received from a customer, there is some specific information that need to be mentioned. Specifically:
- The date of the event, which can be different from the event was reported to the manufacturer.
- The place when the event occurred.
- A description of the event, thus what exactly happened, or a description of the failure or potential failure, etc
- The medical device involved, including all the information needed to properly identify
- Any information related to the involvement of the patient in the specific event and, in case, the consequences of the event on patient, user or any other person involved. If there were consequences, it is often necessary to know whether there was medical intervention on the patient and/or in general the so-called patient outcome.
These are the minimal amount of information which are necessary to open the complaint and perform an initial evaluation of the complaint. Sometimes, this information is not available and it is necessary to contact the customer to collet all the necessary pieces of information.
Reportability Evaluation
We have been extensively talking about the criteria for reportability for US and EU markets, thus we will not go through those requirements again. Here it is just important to highlight that it is essential to collect all the necessary information to perform an adequate evaluation of the reportability of the event.
Technical Investigation
In another article we have been discussed about the techniques for root-cause analysis. In a context of complaint handling, the technical investigation can be quite different depending on the type of events and type of potential failures. However, in general, the technical investigation may contain the following items:
- the root-cause of the event, thus if it is related to a specific failure of the device, use error, or any other reason.
- If for root-cause analysis, it is necessary to evaluate the sample involved in the specific complaint, the results of sample analysis shall be clearly documented or referenced in the complaint record.
- review of the device history record for the device involved in the event, to check if any deviation potentially related to the specific event has been recorded in manufacturing documentation. Sometimes it is necessary to check as well device history record of devices manufactured in the same period of the one involved in the event (for example previous and subsequents lot or serial numbers).
- Complaint data history: it is often necessary to provide an overview of how many similar events have been received within a specific timeframe.
- If medical investigation had to be performed because the event imparted a consequence to a patient, the results shall be included in the investigation of the complaint. Often medical investigation is needed to evaluate the seriousness of the event (serious injury or not) and thus perform the appropriate reportability evaluation.
Risk Assessment Evaluation
It is important, once the root-cause of the event is determined, that a risk assessment is performed, to ensure the risk is still considered acceptable. It is important to remind it is not possible to have unacceptable risks for marketed medical device; once a risk becomes unacceptable, a field action or field safety notice shall be implemented.
Evaluation of the need to implement corrective action
Once the root-cause is determined, the evaluation of the necessity to implement corrective action shall be performed and documented. If no specific actions are taken, appropriate justification shall be documented.
Response to Customer
Response to customers with a summary of the complaint received and the results of the in investigation shall be provided under the following situations:
- the customer specifically requires a response
- a competent authority asks the manufacturer to provide a response
Post-Market Surveillance Training at QualityMedDev Academy
QualityMedDev Academy provides online & self-paced training courses focused on quality and regulatory affairs topics for medical device business. Developed by highly skilled professionals, these trainings offer a great opportunity to increase your competencies over a broad range of regulatory topics.
Do not hesitate to take a look to our course on Complaint Handling and Vigilance Reporting Process .

4EasyReg Documentation on Complaint Management
Considering the importance of the complaint handling process, 4EasyReg presents a series of documents that can be easily downloaded and used to organise your complaint handling process:
- EU Vigilance Reporting Procedure
- Complaint Handling Procedure
- Complaint Handling Template
Moreover, 4EasyReg provides a Post-Market Surveillance Procedure that defines all the requirements associated to PMS activities according to EU MDR 2017/745 and IVDR 2017/746. This includes PMS Plan, PMCF Plan and Report, PUSR, PMS Review, frequency of review and update of PMS processes, and much more. The procedure is a 10-pages word file, fully editable and ready to be amended in order to align the documents with the internal processes of your organization.
Subscribe to 4EasyReg Newsletter
4EasyReg is an online platform dedicated to Quality & Regulatory matters within the medical device industry. Have a look to all the services that we provide: we are very transparent in the pricing associated to these consulting services.
Within our WebShop, a wide range of procedures, templates, checklists are available, all of them focused on regulatory topics for medical device compliance to applicable regulations. Within the webshop, a dedicated section related to cybersecurity and compliance to ISO 27001 for medical device organizations is also present.
As one of the leading online platforms in the medical device sector, 4EasyReg offers extensive support for regulatory compliance. Our services cover a wide range of topics, from EU MDR & IVDR to ISO 13485, encompassing risk management, biocompatibility, usability, software verification and validation, and assistance in preparing technical documentation for MDR compliance.
Do not hesitate to subscribe to our Newsletter!
