Medical device containing substances or tissues derived from animals are important class of devices and they are highly regulated according to specific ISO standards, the ISO 22442 family of standards.
Obviously the general processes applicable for any type of devices are still applicable, such as risk management process, usability and all the standards related to validation activities (for example biocompatibility). However, for this type of devices, specific standards are needed in order to properly apply all the general standards. In fact, considering for example the risk management process, specific risks need to be taken in consideration, for example contamination by bacteria, virus, agents that cause TSE (Transmissible spongiform encephalopathy) or anything else that may cause an undesirable pyrogenic, immunologic or toxicologic reaction.
In the next section we will go through the main requirements associated to ISO 22442 family of standards, with a particular emphasis to ISO 2242-1 related to the application of risk management process for medical devices utilizing animal tissues and related derivates.
We have already been discussing about the risk management process for medical devices, including the requirements of ISO 14971 and ISO 24971 and the link between risk management and post-market surveillance.
ISO 22442-1: Application of Risk Management Process
First of all it is important to mention to the ISO 22442-1 is not a stand-alone standard and can only be used in combination with ISO 14971, as clearly mentioned in the introduction of the standard.
The general risk management process described in the standard can be pictured out in the scheme below :
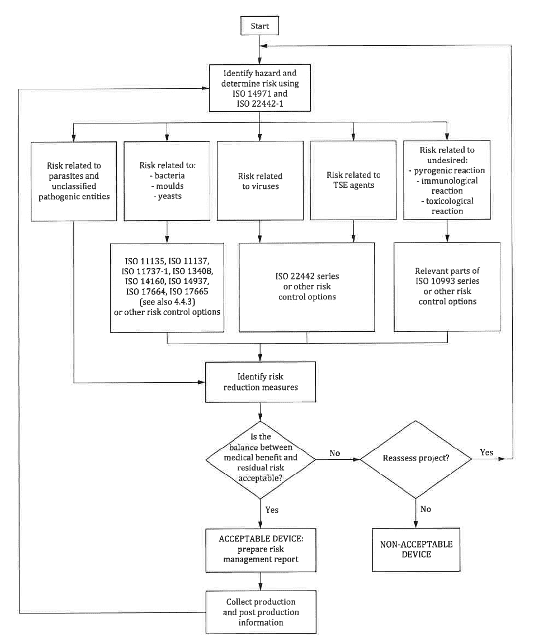
Analysis of the risks for devices containing animal tissues according to ISO 22442-1
Different aspects need to be considered to perform an adequate analysis of the risks. For example, it has to be evaluated if the type of contact between the body and the material derived from animal tissue. Aspects to consider include contact duration, the type of fluid that may come in contact and other similar topics.
Moreover, a careful analysis on the type of materials in contact with or incorporated into the medical device shall be performed. In this case, different factors shall be analysed, such as:

Moreover, the conditions under which the device is supplied also play a fundamental role. In fact, as reported by the standard, variations in the bioburden level of bacteria or other microorganisms in the tissues shall be estimated.
One last point to evaluate is the presence of toxic residues, maybe coming from the manufacturing process; this should also be fully considered from a risk stand point.
Identification of hazards and hazardous situation
The subsequent step is the identification of hazards & hazardous situation, starting from the analysis of the risks performed based on the considerations of the previous section.
Particular attention shall be provided to potential contamination issues, for example by transmissible agents or for the presence of contaminants on the finished material that may cause adverse reactions.
Risk Evaluation
Risks shall be evaluated using a classic method reported in the ISO 14971:2019. A specific risks evaluation shall be performed to address the risks related to transmissible agents.
Risk Control
The main risk control measures for devices containing substances or tissues derived from animals are the compliance with the ISO 22442 family of standards. Other relevant risk control measures are related to compliance to ISO standards related to sterilization or cleaning or risk control options linked to biocompatibility tests (ISO 10993 family of standards).
Residual Risks Evaluation and Acceptability according to ISO 22442-1
The next step in the classic risk management process is the residual risk acceptability, that shall be performed at the level of each single risk.
For risk related to TSE (Transmissible spongiform encephalopathy), there are specific requirements for the acceptability of the associated risk.
In fact, based on the ISO 22442-1, TSE related risks can be considered acceptable under the following conditions:
- The estimation of the residual risk indicates that TSE risks have been reduced as much as possible to an acceptable level
- In a context of a risk/benefit analysis, the medical benefit arising from the intended use of the device is judged to outweigh the residual risk estimate.
Overall Residual Risk Acceptability
One of the last step in the risk management process is related to final considerations on the overall risk acceptability. This evaluation shall typically be performed by evaluating each residual risk after the implementation of the risk control measures and the expected medical benefit of the specific medical device.
Particular attention shall be given to the situation where there are residual risks related to contamination with transmissible agents. Under these circumstances, it shall be evaluated whether a) alternative materials with a better risk/benefit profile may be used b) alternative products with the same intended may be used.
Production and Post-Production Information
For this type of medical devices, in the framework of the collection of information for post-market risk management, it is essential to evaluate any changes in the zoonosis status of the chosen source of animal material. Zoonoses are defined as those diseases and infections naturally transmitted between people and vertebrate animals.
Specific requirements according to EU MDR
The EU MDR 2017/745 provides in the Chapter II of the Annex I of the text, a section specifically dedicated to the requirements for this type of device.
The regulation makes a distinction between:
- Devices utilizing derivatives of tissues or cells of human origin which are non-viable or are rendered non-viable; and
- Devices utilizing tissues or cells of animal origin which are non-viable or are rendered non-viable;
For the first type of devices, the specific applicable requirements can be summirezed as follows:
- The directive 2004/23/EC shall be applied for the donation, procurement and testing of the tissues and cells used within the device
- High attention to safety towards patient, user or any other person shall be paid during the handling of tissues, cells and related derivatives. In particular, specific attention shall be given in regards of viruses and other transmissible agents and the use of validated method for the elimination of potential sources of transmission.
- Traceability system that shall be aligned with the applicable Directive 2004/23/EC and in Directive 2002/98/EC

On the other hand, for devices using tissues and cells of animal origin, the following requirements need to be met:
- Animal source of these materials shall have been subjected to veterinary control that are adapted to the intended use of the tissue. Moreover, the information on the geographical origin of the animals shall be retained.
- Also in this case, safety to patients, users, or other persons shall be the priority. So it is important that sourcing, processing, preservation, testing and handling of tissues, cells and substances of animal origin, or their derivatives, shall be carried out in a way to preserve safety of the device when used on a patient. Also in this case, specific attention shall be given in regards of viruses and other transmissible agents and the use of validated method for the elimination of potential sources of transmission.
- The regulation (EU) No 722/2012 shall apply in case of devices manufactured utilising tissues or cells of animal origin, or their derivatives.
In general we can say there is an alignment between MDR requirements and the ISO 22442 family of standards.
Subscribe to 4EasyReg Newsletter
4EasyReg is an online platform dedicated to Quality & Regulatory matters within the medical device industry. Have a look to all the services that we provide: we are very transparent in the pricing associated to these consulting services.
Within our WebShop, a wide range of procedures, templates, checklists are available, all of them focused on regulatory topics for medical device compliance to applicable regulations. Within the webshop, a dedicated section related to cybersecurity and compliance to ISO 27001 for medical device organizations is also present.
As one of the leading online platforms in the medical device sector, 4EasyReg offers extensive support for regulatory compliance. Our services cover a wide range of topics, from EU MDR & IVDR to ISO 13485, encompassing risk management, biocompatibility, usability, software verification and validation, and assistance in preparing technical documentation for MDR compliance.
Do not hesitate to subscribe to our Newsletter!
