The part of the Code of Federal regulation 21 CFR Part 820 is the section related to the requirements for Quality Management System of medical device manufacturers that want to sell medical products in the United States. It is composed by the official section of the regulation – indeed 21 CFR Part 820 – and the preamble of the regulation, where the agency provides interpretation of the requirements, considering also the public comments that have been received.
Every manufacturers that is distributing their devices in the United States need to have a Quality Management System fully established according to FDA requirements. Similarly to the requirements of ISO 13485:2016, the quality system shall be commensurate with:
- risks presented by the device
- complexity with the device and the manufacturing operations
- size and complexity of the organization
Quality Management Subsystems As per 21 CFR 820
In the scheme below, we have a schematic representation of the quality management subsystems according to 21 CFR 820:
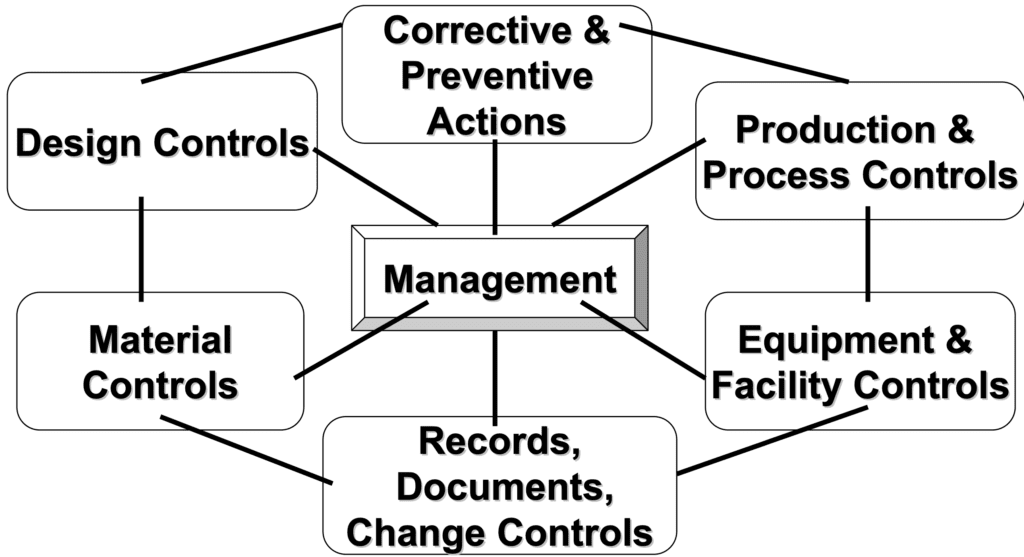
We will now provide a full overview of the main requirements associated to 21 CFR 820, considering all the subsystem mentioned in the scheme reported above. This article will not cover all the requirements of the 21 CFR 820 regulations; only a selection of the requirements will be discussed.
The requirements that will not be specifically discussed in this article will be covered by other articles in the QualityMedDev blog.
Management Requirements according to 21 CFR 820
Within the Quality System regulation, management has very specific responsibilities. Firstly, it has the responsibility to establish a quality policy. In fact, the quality policy shall be established by management with executive responsibility and must be understood, implemented, and maintained at all levels of the organization.
Moreover, the management has responsibilities related to the maintenance an adequate organizational structure with specific authorities and interrelation of personnel and sufficient resources. Furthermore, management shall also appoint a management representative.
One of the method used by management to keep the quality system under control is the Management Review. In fact, management with executive responsibility shall review the suitability and effectiveness of the quality system:
- At defined intervals
- With sufficient frequency
- According to established procedures
Quality Audit and 21 CFR 820
In order to ensure compliance of the quality system to 21 CFR 820 FDA regulation, procedures and specific plans for quality audit shall be performed. Audits shall be performed by individuals with no direct responsibilities in the area which is audited. An audit report shall be prepared with the results of the audit and any non-conformity shall be corrected through the so-called CAPA process.
HR Processes
It is of key importance to establish procedures for identifying training needs and to ensure personnel are adequately trained. Moreover, documentation related to education, background, training, and experience of the personnel shall be kept in the quality system.
Design Controls and FDA 21 CFR 820
One of the most important process within a Quality System is design control. As specified by the agency, the design control requirements are not intended to apply to development of concepts and feasibility studies. Moreover for investigational device exemption (IDE) device , the FDA has amended the IDE regulation, reaffirming that an IDE device is exempted from complying with the GMP’s, with the exception of the requirements related to “Design Controls.”
We have already been discussing about the main steps of the design control process, so here in this article we will grow through them quite quickly.
Design and Development Planning
The Design and Development Plan has different purposes, such as:
- Define responsibility for implementation.
- Identify and describe interfaces between different groups or activities.
- Review, update, and approve plans as design and development evolves.
Design Inputs
In the design inputs phase, the requirements of the device shall be defined in order to address the intended use of the device. Incomplete, ambiguous, or conflicting requirements shall be addressed and resolved.
Design Outputs
Design outputs shall be defined in a way that it allows the evaluation of the fulfilled of the design inputs. Moreover, design outputs essential for the proper functioning of device shall be properly identified. It is essential to document, review, and approve design outputs before release of the device. The design outputs are the basis for the preparation of the Device Master Record.
Design Review
The Design review can be considered a documented, comprehensive, systematic examination to
- Evaluate adequacy of the design requirements
- Evaluate capability of the design to meet requirements
- Identify problems
It is essential to ensure that formal reviews of design results are planned and conducted at specific stages of the design process. The results of the design review shall be documented in the Design History File (DHF).
Design Verification, Validation and Design Transfer
The section related to Design verification and Validation has been extensively treated in a different article at QualityMedDev, thus we’ll not speak more in this article.
The design transfer, instead, shall ensure the device design is correctly translated into production specifications.
Purchasing Controls
It is of fundamental importance to establish and maintain procedures to ensure that all purchased or otherwise received product and services conform to specified requirements. Moreover, all the suppliers, subcontractors and consultants shall be evaluated using a risk-based approach.
Furthermore, purchasing data/documents that describe or reference specified requirements (including notification of change agreements) shall be maintained.
Process Validation
The requirements for Process Validation are define din the section of 21 CFR 820. The validation activities for a specific process is needed to ensure the outcome of the process is consistent over the time. The processes to be validated are the ones for which the results cannot be fully verified by subsequent inspection and test. We have been already talking about the approaches for Process Validation in another article of QualityMedDev. In general, the standard approach for Process Validation is based on three different phases which are called:
- IQ – Installation Qualification
- OQ – Operational Qualification
- PQ – Performance Qualification
Conclusions
In conclusion, we have been discussing some of the requirements related to 21 CFR 820 FDA regulation. As mentioned before, this article did not cover all the requirements of the 21 CFR 820 regulations; only a selection of the requirements has been discussed. Moreover, some specific topics, like CAPA management, Change Control and Process Validation, have been already discussed in other articles.
